CHAPITRE I : Rappels bibliographiques
Bien que le zinc soit un composé assez peu abondant dans la nature, sa non-toxicité et sa simplicité d’isolement font que ce métal et ces sels sont peu coûteux et sont à priori intéressants pour la synthèse organique(1). Effectivement, à l’heure actuelle les dérivés organozinciques sont des réactifs couramment utilisés pour la synthèse de squelettes carbonés(). Cependant, malgré leur découverte par Frankland dès 1849 (2), ces composés ont été laissés de coté pendant plus de 100 ans au profit des dérivés organomagnésiens et des organolithiens, beaucoup plus réactifs. Le regain d’intérêt des dérivés organozinciques est dû justement à leur faible réactivité qui permet de tolérer sur un même squelette carboné à la fois la fonction organométallique et des fonctions très réactives : cétones, esters, nitriles, etc…, lesquelles sont incompatibles avec la présence de liaison C-Mg ou C-Li si ce n’est pour ces derniers à très basse température (≤-100°C). Surtout un grand nombre de travaux récents ont mis en évidence que l’on pouvait augmenter sélectivement la réactivité des ces zinciques vis à vis d’un substrat donné à l’aide d’une catalyse homogène par un complexe d’un métal de transition judicieusement choisi. On peut donc avoir en principe accès à des molécules poly fonctionnelles d’un grand intérêt, à conditions bien sûr d’être capable de synthétiser efficacement le réactif organométallique que l’on peut qualifier de « doux » du fait de sa stabilité. A l’heure actuelle, il y a 2 grandes méthodes chimiques pour préparer les organozinciques. La plus directe (3)analogue à la réaction de Grignard, met en jeu la réaction d’un halogénure organique RX avec du zinc activé.
![]() RX + Zn* RZnX
RX + Zn* RZnX
La deuxième (4)consiste d’abord à former un organométallique usuel (magnésien ou lithien) RM auquel on fait subir une réaction de transmétallation par un sel de zinc.
![]() RM
+ ZnX2 RZnX
+ MX
RM
+ ZnX2 RZnX
+ MX
Cette méthode n’a qu’un intérêt limité puisqu’elle ne permet généralement l’accès qu’à des dérivés dépourvus de groupes fonctionnels réactifs.
Nous avons choisi de nous intéresser à la synthèse de dérivés organozinciques aromatiques et hétéroaromatiques, lesquels sont difficilement accessibles voir inaccessibles à partir de dérivés bromés et a fortiori des dérivés chlorés si le noyau aromatique ou hétéroaromatique est substitué par des groupes réactifs cités précédemment. Ces derniers dérivés sont des réactifs très intéressant du fait de leur compatibilité fonctionnelle. En présence d’un catalyseur approprié (5)(Schéma 1), il est possible à partir de ces organozinciques aromatiques d’accéder à des biaryles dissymétriques fonctionnalisés(6), à des 2-arylpyridines(7), à des oléfines fonctionnalisées(8), ou encore à des cétones dissymétriques(9) ; ces réactions mettent en œuvre une catalyse homogène par des complexes de Pd(0), Ni(0), Cu(I), ou encore se font en transformant par réaction de transmétallation l’organozincique en d’autres espèces organométalliques plus réactives comme par exemple des dérivés cuprozinciques «Ar-CuZnX2 ».
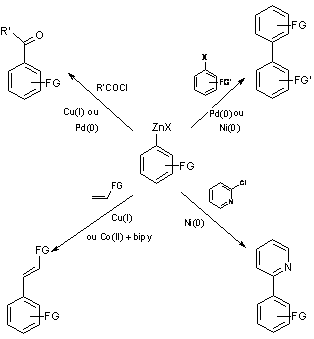
Schéma 1
Outre les voies chimiques précédentes des méthodes par electrosynthèse ont aussi été proposées pour conduire aux organozinciques aromatiques à partir des halogénures aromatiques
Nous présenterons tout d’abord les différentes approches chimiques qui ont été décrites.
1. Formation des organozinciques aromatiques et hétéroaromatiques par voie chimique
1.1. Par réaction de transmétallation
La première méthode nécessite le passage par un organomagnésien ou un organolithien suivi d’une réaction de transmétallation avec un sel de zinc (équation 1).
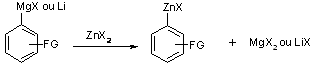
Equation 1
La formation du magnésien ou du lithien ne pose pas de problème lorsque le noyau aromatique est porteur de groupements electrodonneurs.
Par contre, la synthèse de réactifs de Grignard aromatiques ou
hétéroaromatiques porteurs de groupements électroattracteurs s’avère très
difficile. Récemment, Cahiez et Knochel(10)
ont mis au point une synthèse d’organomagnésiens aromatiques et hétéroaromatiques
porteurs de groupements électroattracteurs : ester, amide, nitrile à –10
ou –40°C entre un iodure ou un bromure aromatique et l’isopropylmagnésium
(équation 2).
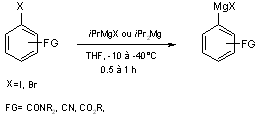
Equation 2
La transformation en zincique correspondant se fait par réaction du magnésien aromatique avec un halogénure de zinc (ZnBr2 ou ZnCl2).
De même, à très basse température (-100°C), il est possible d’accéder à des organolithiens aromatiques porteurs de groupements réactifs (équation 3) et par conséquent aux zinciques correspondants(4).
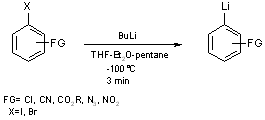
Equation 3
Bien qu’il soit maintenant possible d’obtenir des aryllithiens et des arylmagnésiens fonctionnalisés à partir des iodures aromatiques et parfois des bromures, les réactions mettent en jeu plusieurs étapes et se font à basse , voir très basse températures.
1.2. Echange halogène-métal
Méthode utilisant le zinc
actif noté Zn* (zinc de Rieke)
![]()
Rieke prépare un zinc très réactif (Zn*) par réduction en milieu THF, d’un
halogénure de zinc par le naphtalène sodium ou par le naphtalène lithium(3).
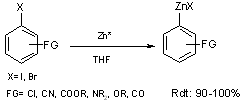
Ce zinc ainsi préparé (obtenu sous forme d’une boue), réagit directement avec
différents iodures et bromures aromatiques porteurs de fonctions réactives(11) (équation 4).
Equation 4
Les iodures aromatiques réagissent rapidement avec le zinc actif à température ambiante, alors qu’avec des bromures aromatiques il faut se placer plusieurs heures à reflux. Par contre, il n’est pas possible de synthétiser le zincique d’un chlorure aromatique.
Ce zinc actif ne réagit pas directement avec le 3-bromothiophène. Il est par contre actif vis à vis du 3-iodothiophène. La transformation du 3-bromothiophène en zincique se fait donc par la suite des réactions données par l’équation 5.
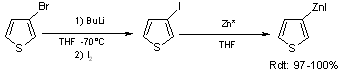
Equation 5
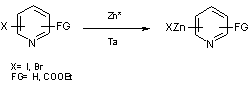
Le zinc de Rieke a également été utilisé par Yamanaka et coll. pour synthétiser
les organozinciques dérivés d’iodo ou bromo- pyridines.(13) (équation 6).
Equation 6
La méthode développée par Rieke semble efficace mais très aléatoire, en effet le zinc actif est délicat à obtenir et surtout sa réactivité est peu reproductible dépendant des conditions de sa conservation. Il ne nous semble donc pas que cette méthode puisse être exploitée à grande échelle, bien que ce zinc soit maintenant un produit commercial, de même que les organozinciques aromatiques préparés selon cette voie.
A partir de dérivés iodés
Majid et Knochel (14) ont montré que les iodures porteurs de groupements ester, nitrile, chlorure ou encore cétone, peuvent être converti en organozinciques correspondants par insertion directe du zinc en poudre, activé par réaction avec du dibromoéthane dans la liaison C-I en milieu solvant polaire DMF ou DMAC à des température de l’ordre de 25 à 55°C (équation 7).
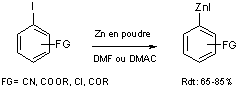
Equation 7
Ce procédé a été étendu à des composés hétéroaromatiques tel que les 2-iodothiophènes ; par exemple le zincique du 2-iodothiophène est obtenu en 1.5 heures à température ambiante avec un rendement de 60%.
Enfin, une dernière voie d’accès aux organozinciques aromatiques à partir des iodures correspondants consiste à utiliser une irradiation ultrasonique pour activer du zinc en poudre (équation 7)
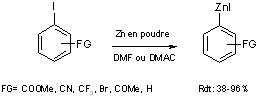
Equation 7
Cette irradiation ultrasonique est indispensable pour obtenir l’organozincique à partir des iodures substitués en para et méta de l’iode. Si les groupements fonctionnels sont portés en ortho de l’iode seul le temps de réaction est diminué du fait du traitement aux ultrasons.
Ces deux méthodes donnent de très bons résultats, mais elles nécessitent l’utilisation de composés iodés qui sont plus onéreux et beaucoup plus difficiles d’accès que les dérivés bromés ou chlorés correspondant.
2. Synthèse des organozinciques aromatiques et hétéroaromatiques par voie électrochimique
Au laboratoire les organozinciques aromatiques peuvent être synthétisés à l’aide du procédé à anode consommable, développé il y a une vingtaine d’années par J. Périchon. Les réactions électrochimiques sont assistées soit par une catalyse par un complexe du nickel soit par une catalyse par un halogénure de cobalt. Dans le cas ou le nickel est le catalyseur le milieu solvant est le diméthylformamide. Avec le cobalt, les réactions ont lieu en milieu diméthylformamide ou acétonitrile associé ou non dans le cas de ce dernier solvant à de la pyridine.
2.1. Catalyse par le nickel
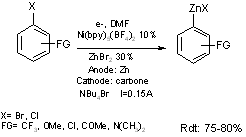
Le premier procédé de synthèse des organozinciques aromatiques (6b)(décrit à l’origine par S. Sibille et J.
Périchon) utilise le procédé à anode consommable, il s’effectue dans une
cellule à compartiment unique munie d’un barreau métallique de zinc qui joue le
rôle d’anode, et d’une cathode cylindrique en fibre de carbone. Le catalyseur
utilisé est un complexe du nickel : Ni(bpy)5(BF4)2.
La réaction a lieu dans le solvant diméthylformamide, en présence de ZnBr2
, et d’un électrolyte indifférent NBu4Br qui assure
une bonne conductivité au milieu (équation 8). Cette réaction permet d’accéder
à des organozinciques aromatiques, porteurs de groupes attracteurs ou donneurs
à partir d’halogénures chlorés ou bromés. Cependant, cette synthèse conduit
dans tous les cas à la formation de 20 à 25% de produits de réduction de
l’halogénure initial, c’est à dire le produit d’hydrogénation ArH et à 5 à 10%
de dimère ArAr.
Equation 8
Les organozinciques ainsi obtenus peuvent être couplés avec divers électrophiles en présence d’un catalyseur approprié : par exemple avec la cyclohex-2-énone , avec le chlorure d’allyle, ou avec divers iodés aromatiques. Les rendements obtenus sont de moyens à bons, quel que soit le groupement fonctionnel porté par le noyau aromatique.
Afin d’optimiser cette méthode (principalement diminuer la quantité de bipyridine) et de l’étendre, C. Gosmini a mis au point de nouvelles conditions expérimentales. Le complexe Ni(bpy)5(BF4)2 a été remplacé par le complexe NiBr2bpy, et l’anode de zinc remplacé par une anode de magnésium ce qui nécessite la présence de ZnBr2 en quantité stœchiométrique(7).
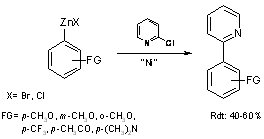
Les différents organozinciques aromatiques ainsi formés ont pu être couplés
avec des 2-halogénopyridines (équation 9) sans qu’il soit nécessaire d’ajouter
un catalyseur . Le complexe du nickel présent dans le milieu et qui a servi à
la synthèse de l’organozincique sert de catalyseur à la réaction de couplage.
Equation 9
Cette méthode a aussi permis de synthétiser pour la première fois directement le zincique du 3-bromothiophène avec un très bon rendement de 80%(16)(équation 10). Comme nous l’avons signalé précédemment, à partir du bromure, aucune méthode décrite dans la littérature ne permettait l’accès direct à ce composé.
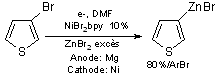
Equation 10
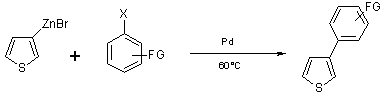
Le zincique du 3-bromothiophène a ensuite été couplé au palladium avec divers
halogénures aromatiques, ou avec des 2-halogénopyridine comme décrit
précédemment.(7, 16)
Rdt : 40-72%
Equation 11
Le procédé électrochimique faisant intervenir des complexes de nickel apporte déjà des améliorations par rapport aux procédés chimiques décrits dans la littérature; d’une part en ce qui concerne la nature des groupes fonctionnels portés par le noyau aromatique, d’autre part car il permet d’utiliser les chlorures aromatiques comme réactif de départ ce qui est complètement original. Le mécanisme de cette réaction met en œuvre le passage par un dérivé s-arylnickel obtenu par réaction entre le nickel zéro valent électrogénéré et l’halogénure aromatique. Le composé s-arylnickel réagit par transmétallation avec les ions zinc présents en solution, et formés par oxydation de l’anode (équation 12)
![]()